chemical properties of group 17 elements
Chemical properties of group 17 elements
Oxidising power
The order of oxidising power is \[\ce{F2}>\ce{Cl2}>\ce{Br2}>\ce{I2}\] (oxidising power decreases down the group), which means that a halogen higher in the group can oxidise the ions of one lower down. For example, chlorine can oxidise the bromide ions: \[\ce{Cl2(aq) + 2 KBr(aq) -> 2 KCl(aq) + Br2(aq)}\]. Such a reaction is also known as a displacement reaction.
Reactions of hydrogen halides
The reactivity of the halogens decreases as we go down the group.
The general form of the equation is: \[\ce{X2(g) + H2(g) -> 2 HX(g)}\].
- Fluorine combines explosively with hydrogen even in the cold and dark
- Chlorine and hydrogen explode if exposed to sunlight or a flame. Alternatively, it will combine more peacefully if you light a jet of hydrogen and then lower it into a gas jar of chlorine.
- Bromine vapour and hydrogen combine with a mild explosion with the presence of a flame
- Iodine and hydrogen only combine partially (reversible reaction) even on constant heating: \[\ce{I2 + H2 <=> 2 HI},\,\Delta H_{f}^{\circ}\,\ce{HI}=+26.5\text{kJmol}^{-1}\]. See enthalpy of formation.
Thermal stability of hydrogen halides
The thermal stability of hydrogen halides decrease down the group as the bond energies between the hydrogen and the halogen decreases. This is due to the increasing atomic radius of halogen atoms which increases the distance between the nucleus and electrons thus decreasing the effective nuclear charge experienced by the electrons.
\[\ce{HF}\] and \[\ce{HCl}\] are stable to heat and do not decompose when heated at normal laboratory temperatures while \[\ce{HBr}\] decomposes to a small degree. For \[\ce{HI}\], the reaction is reversible thus an equilibrium is established \[\ce{2 HI <=> H2 + I2}\].
Reducing properties of halide ion
\[\ce{F}\] and \[\ce{Cl}\]
Let's use concentrated sulfuric acid, \[\ce{H2SO4}\], as our oxidising agent. The reason for the high concentration is due to the Collision theory.
Concentrated \[\ce{H2SO4}\] isn't a strong enough oxidising agent to oxidise fluoride or chloride ions (doesn't mean there's no reaction happening, just that oxidation numbers are unchanged). The most we'll observe is the release of hydrogen halide gas such as \[\ce{HCl(g)}\] (as it's boiling point is 19.5C and -85C respectively).
\[\ce{NaCl(s) + H2SO4(aq) -> HCl(g) + NaHSO4(aq)}\] and \[\ce{NaF(s) + H2SO4(aq) -> HF(g) + NaHSO4(aq)}\]
The process would go like this:
- Breaking the attractions between the ions in the crystalline solid halide structure such as the \[\ce{NaF}\] structure (energy required decreases from \[\ce{F}\] to \[\ce{I}\])
- Removing an electron from the halide ion (e.g. \[\ce{Cl- -> Cl}\]), which requires an input of energy equivalent to it's first ionisation energy (energy required also decreases from \[\ce{F}\] to \[\ce{I}\])
- Recovering a tiny portion of the energy when the halogen atoms forms halogen molecules (e.g. from \[\ce{2F}\] to \[\ce{F2}\])
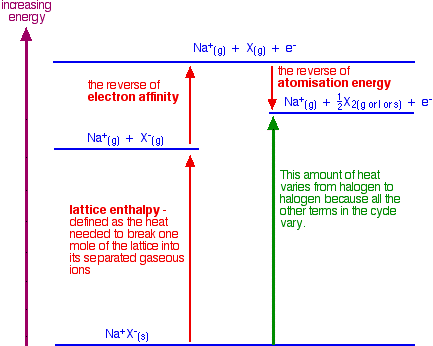
Which would explain the trend of an increase in size of ions (in a simple and slightly inaccurate manner).
- The bigger the halide ion (going down the group), the easier it is for it to lose an electron, thus making it a better reducing agent
- The bigger the halogen molecule (\[\ce{X2}\]), the ability for it to be an oxidising agent decreases, as the nucleus exerts a weaker attraction on the valence electrons as the atomic radius increases.
The following \[\Delta H_{f}^{\circ}\] values are retrieved from the NIST-JANAF Thermochemical Tables.
To determine why concentrated sulfuric acid isn't strong enough, let's take the enthalpy change for the reaction \[\ce{NaCl(s) + H2SO4(aq) -> HCl(g) + NaHSO4(aq)}\] as an example:
Assuming the enthalpy of formation (\[\text{kJmol}^{-1}\] under the standard conditions) is approximately, \[\Delta H_{f}^{\circ}(\ce{NaCl(s)})=-411.1\], \[\Delta H_{f}^{\circ}(\ce{H2SO4(aq)})=-814.0\], \[\Delta H_{f}^{\circ}(\ce{HCl(g)})=-92.3\], \[\Delta H_{f}^{\circ}(\ce{NaHSO4(aq)})=-1125.5\].
Which as shown here, is ever so slightly endothermic (requires input of heat to react at a temperature of 298.15K), thus no significant chemical changes will happen at room temperature, however, with heat added, \[\ce{HCl}\] (colourless) gas will be released steadily.
The reason why \[\ce{HCl(g)}\] does not get further oxidised into \[\ce{Cl2}\] (through the reaction of \[\ce{2HCl(g) + H2SO4(aq) -> 2H2O(l) + SO2(g) + Cl2(g)}\]) can too be explained with the enthalpy changes.
Assuming the enthalpy of formation (at the same conditions as the above), \[\Delta H_{f}^{\circ}(\ce{H2O(l)})=-285.8\], \[\Delta H_{f}^{\circ}(\ce{SO2(g)})=-296.8\], \[\Delta H_{f}^{\circ}(\ce{Cl2(g)})=0\].
As seen here, the reaction is much more endothermic, thus this reaction is seen as energetically unviable.
Additionally, fluoride ions are very difficult to oxidise to fluorine. However, the reasons aren't the usual ones you would expect. Fluoride ions are so small that the electrons feel an abnormal amount of repulsion from each other. This actually outweighs the effect of their closeness to the nucleus and makes them easier to remove than you might expect. On the flip side, due to the small size of the fluoride ion, the positive and negative ions are very close together in the crystal lattice structure thus strongly attracted to each other.
\[\ce{Br}\] and \[\ce{I}\]
For bromide ions, some of the hydrogen bromide, \[\ce{HBr}\], formed will get oxidised into \[\ce{Br2}\]. First, the \[\ce{H2SO4}\] acts as an acid, donating \[\ce{H+}\] to \[\ce{Br-}\], \[\ce{NaBr(s) + H2SO4(aq) -> HBr(g) + NaHSO4(aq)}\].
Assuming the enthalpy of formation, \[\Delta H_{f}^{\circ}(\ce{NaBr(s)})=-361.4\], \[\Delta H_{f}^{\circ}(\ce{HBr(g)})=-36.4\], \[\Delta H_{f}^{\circ}(\ce{Br2(g)})=0\].
Now some of the \[\ce{HBr}\] gets oxidised into \[\ce{Br2}\] (brown-ish colour) through the reaction \[\ce{2HBr(g) + H2SO4(aq) -> 2H2O(l) + SO2(g) + Br2(g)}\].
Although it may seem like the initial reaction for \[\ce{NaBr}\] is slightly more endothermic than the initial reaction for \[\ce{NaCl}\], we have to keep in mind that:
- The subsequent oxidation of \[\ce{HBr}\] into \[\ce{Br2}\] should be exothermic (but our calculation indicates a slightly endothermic reaction), which compensates for the initial energy input.
- Our values for the enthalpy of formation may be slightly incorrect as the NIST-JANAF database dates back to 1998 (but we'll still use it for calculation purposes).
However, our point still stands as the \[\Delta H_{\text{reaction}}^{\circ}\] for \[\ce{HBr}\] to be oxidised into \[\ce{Br2}\] is far lower than oxidising \[\ce{HCl}\] into \[\ce{Cl2}\].
Similarly, for iodide ions, we have our initial reaction of \[\ce{NaI(s) + H2SO4(aq) -> HI(g) + NaHSO4(aq)}\]. However, now we have three reaction pathways:
- \[\ce{2HI(g) + H2SO4(aq) -> I2(g) +SO2(g) + 2H2O(l)}\]
- \[\ce{6HI(g) + H2SO4(aq) -> 3I2(g) + S(s) + 4H2O(l)}\]
- \[\ce{8HI(g) + H2SO4(aq) -> H2S(g) + 4H2O(l) + 4I2(g)}\]
The observations we would most likely see would be purple \[\ce{I2}\] vapour, the pungent \[\ce{SO2}\] and \[\ce{H2S}\] gas and pale yellow solid \[\ce{S}\].
Halide ions test with silver nitrate, \[\ce{AgNO3}\]
Unlike most ionic halides, silver halides are insoluble in water with the exception of silver fluoride, \[\ce{AgF}\]. When acidified silver nitrate solution (\[\ce{AgNO3}\] combined with dilute \[\ce{HNO3}\] which prevents the formation of precipitates from other ions in the solution) is added to the halide solution, precipitate is formed.
- \[\ce{Ag+(aq) + F-(aq) -> AgF(aq)}\], soluble, thus no precipitate
- \[\ce{Ag+(aq) + Cl-(aq) -> AgCl(s)}\], white precipitate of silver cholride which soluble in dilute ammonia solution, \[\ce{NH3}\]
- \[\ce{Ag+(aq) + Br-(aq) -> AgBr(s)}\], cream precipitate of silver bromide which is soluble in concentrated ammonia solution
- \[\ce{Ag+(aq) + I-(aq) -> AgI(s)}\], pale yellow precipitate of silver iodide which is also insoluble in both dilute/concentrated ammonia solution

It is to note that all ionic compounds, even those considered "insoluble," dissolve slightly in water, producing a small concentration of ions, thus there is no such thing as an absolutely insoluble ionic compound. The solubility of sparingly soluble ionic compounds is quantified using the solubility product constant, denoted as \[K_{sp}\] (a predefined constant).
For silver halides, \[\ce{AgX}\], where \[\ce{X}\] represents \[\ce{Cl-}\], \[\ce{Br-}\] and so on, it's solubility product expression is then represented as \[K_{sp}=[\ce{Ag+ _{(aq)}}][\ce{X- _{(aq)}}]\], where each square bracket represents it's concentration in \[\text{moldm}^{-3}\]. It's ionic product on the other hand is the product of the actual ion concentrations in solution. If the ionic product > \[K_{sp}\], then the solution is supersaturated, and excess ions precipitate until equilibrium is restored (ionic product = \[K_{sp}\]). If the ionic product is less than \[K_{sp}\], no precipitate forms as the solution can hold more dissolved ions.
\[K_{sp}\] values for silver halides:
| Compound | \[K_{sp}\] \[\left(\text{mol}^{2} \text{dm}^{-6}\right)\] |
|---|---|
| \[\ce{AgCl}\] | \[1.8\times 10^{-10}\] |
| \[\ce{AgBr}\] | \[7.7\times 10^{-13}\] |
| \[\ce{AgI}\] | \[8.3\times 10^{-17}\] |
You can see that the \[K_{sp}\] values are very small, which makes it very insoluble (will form a lot of precipitate), and becomes even less soluble as you go down the group.
Now, when ammonia solution is added into the silver halide solution, the ammonia, reacts with silver ions to form diamminesilver(I) ion through the reaction \[\ce{Ag+(aq) + 2NH3(aq) <=> [Ag(NH3)2]+(aq)}\]. Although this is a reversible reaction, but the complex ion is very stable, and the position of equilibrium lies well to the right. This reaction reduces the concentration of free \[\ce{Ag+}\] ions in solution because they are either tied up as in the form of \[\ce{AgX}\] or \[\ce{Ag(NH3)2+}\]. However, this also causes the ionic product to reduce significantly (as ionic product = \[[\ce{Ag+ _{(aq)}}][\ce{X- _{(aq)}}]\], and a decrease in free \[\ce{Ag+}\] means a decrease in ionic product), making the ionic product < \[K_{sp}\], thus the precipitate will dissolve to release \[\ce{Ag+}\] to re-establish equilibrium (achieve ionic product = \[K_{sp}\]). Which matches what we observe when we mix silver chloride/bromide solution with ammonia solution.
The reason why \[\ce{AgI}\] does not dissolve even with ammonia solution added is due to it's incredibly low \[K_{sp}\]. Even concentrated ammonia solution is insufficient in lowering the ionic product below \[K_{sp}\].
Reactions of chlorine
The reaction between chlorine and cold dilute sodium hydroxide solution is \[\ce{2NaOH + Cl2 -> NaCl + NaClO + H2O}\]. \[\ce{NaClO}\] is known as sodium chlorate (I), which is a powerful oxidising agent and is capable of oxidising many coloured substances to being colourless. Thus, the right hand side of the reaction is also, commonly known as bleach.
Oxidation states (an example of a disproportionation reaction):
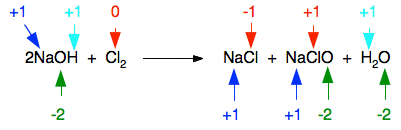
The reaction between chlorine and hot concentrated sodium hydroxide solution is also a disporportionation reaction: \[\ce{6NaOH + 3Cl2 -> 5NaCl + NaClO3 + 3H2O}\]. \[\ce{NaClO3}\] is known as sodium chlorate (V), the V meaning \[\ce{Cl}\] has a +5 charge.
References
- Malcolm W. Chase, Jr. NIST-JANAF Thermochemical Tables. Washington, DC: New York: American Chemical Society; American Institute of Physics for the National Institute of Standards and Technology, 1998.
- Billon, Sophie and Vieillard, Philippe. "Prediction of enthalpies of formation of hydrous sulfates" American Mineralogist, vol. 100, no. 2-3, 2015, pp. 615-627.