Aufbau principle
Aufbau principle
The Aufbau principle, from the German word Aufbauprinzip, which means "building-up principle" states that in the ground state of an atom or ion, electrons first fill subshells of the lowest available energy, then fill subshells of higher energy. The order is shown below:
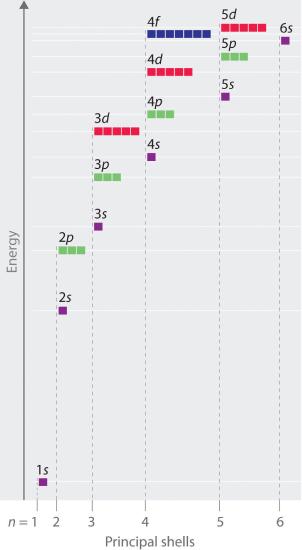
Do note that the energy levels of the orbitals are not constant, they vary as they are filled with electrons. See here for an explanation with experimental evidence.
An easy way to remember this is by remembering that generally subshells with a lower \[n+\ell\] value generally gets filled first. In general, subshells with the same \[n+\ell\] value have similar energies, but the \[\ell=0\] orbitals are exceptional: their energy levels are appreciably far from those of their \[n+\ell\] group and are closer to those of the next \[n+\ell\] group, which allows exceptions to the Aufbau principle.
The configuration is often abbreviated by writing only the valence electrons explicitly, while the core electrons are replaced by the symbol for the last previous noble gas in the periodic table, placed in square brackets. For phosphorus, 1s22s22p63s23p3, the last previous noble gas is neon, so the configuration is abbreviated to [Ne] 3s23p3.
The behaviour of electrons filling of orbitals is further elaborated by other principles such as Pauli's Exclusion principle and Hund's rule.
Despite electrons being filled according to the orbitals with lowest energy levels, during ionisation, electrons are removed starting from the outermost orbital as the electrons in outermost orbital has the highest energy level. So for example Thallium, [Xe] 6s24f145d106p1 (that's according to how the orbitals are filled), when ionised, electrons leave in order 6p, 6s, 5d, 4f, etc.
Shortfalls
The Aufbau principle isn't rigorous because it's based upon the approximation that the electron-electron interaction can be averaged into a mean field. It's only works for most elements but not all because you can't just add up electronic energies using a shell-by-shell, electron-by-electron approach. As soon as you have two or more electrons in close quarters, they interact with each other in ways that cannot be described by simple electrostatic laws because of the quantum mechanical nature of the electrons.
For example, the anomalous configuration of Cr (3d5 4s1) is typically explained as being due to "half-filled subshell stability". This is wrong for several reasons. First of all there is nothing especially stable about half-filled subshells. Secondly it does not explain the fact that many second transition series elements show anomalous configurations even though they do not possess half-filled subshells. The possession of a half-filled subshell (henceforth abbreviated as hfss) is neither necessary nor sufficient for there to be an anomalous configuration. There are atoms that have hfss but do not have anomalous configurations and there are atoms that have anomalous configurations but do not have hfss. Electronic levels have to be found using quantum calculations taking into account electron-electron interactions (not to mention spin orbit coupling). Therefore there is no simple or rational explanation for the exceptions.
For chromium, it turns out the 4s and 3d orbitals are virtually degenerate. This means that the energy required to pair two electrons in the 4s orbital is more than the energy required to move up and put an electron in the 3d orbital. No need to invoke electron exchange energy here. For Cu, the 3d orbital is simply lower in energy than the 4s and lower than the pairing energy so it fills up first.
See this and this for a more detailed explanation. Also this.